Pipes and Filters
Overview
Teaching: 25 min
Exercises: 10 minQuestions
How can I combine existing commands to do new things?
Objectives
Redirect a command’s output to a file.
Process a file instead of keyboard input using redirection.
Construct command pipelines with two or more stages.
Explain what usually happens if a program or pipeline isn’t given any input to process.
Explain Unix’s ‘small pieces, loosely joined’ philosophy.
Now that we know a few basic commands,
we can finally look at the shell’s most powerful feature:
the ease with which it lets us combine existing programs in new ways.
We’ll start with a directory called molecules
that contains six files describing some simple organic molecules.
The .pdb extension indicates that these files are in Protein Data Bank format,
a simple text format that specifies the type and position of each atom in the molecule.
$ ls molecules
cubane.pdb ethane.pdb methane.pdb
octane.pdb pentane.pdb propane.pdb
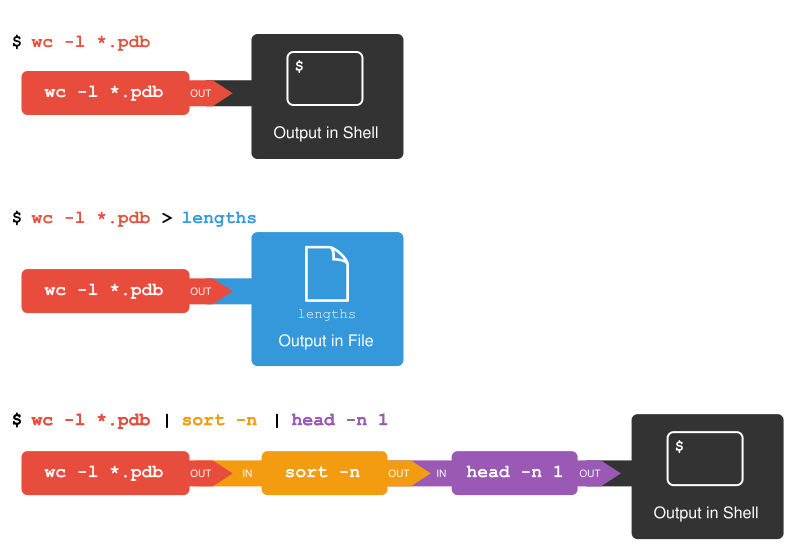
Let’s go into that directory with cd and run the command wc *.pdb.
wc is the “word count” command:
it counts the number of lines, words, and characters in files (from left to right, in that order).
The * in *.pdb matches zero or more characters,
so the shell turns *.pdb into a list of all .pdb files in the current directory:
$ cd molecules
$ wc *.pdb
20 156 1158 cubane.pdb
12 84 622 ethane.pdb
9 57 422 methane.pdb
30 246 1828 octane.pdb
21 165 1226 pentane.pdb
15 111 825 propane.pdb
107 819 6081 total
If we run wc -l instead of just wc,
the output shows only the number of lines per file:
$ wc -l *.pdb
20 cubane.pdb
12 ethane.pdb
9 methane.pdb
30 octane.pdb
21 pentane.pdb
15 propane.pdb
107 total
We can also use -w to get only the number of words,
or -c to get only the number of characters.
Which of these files is shortest? It’s an easy question to answer when there are only six files, but what if there were 6000? Our first step toward a solution is to run the command:
$ wc -l *.pdb > lengths.txt
The greater than symbol, >, tells the shell to redirect the command’s output
to a file instead of printing it to the screen. (This is why there is no screen output:
everything that wc would have printed has gone into the
file lengths.txt instead.) The shell will create
the file if it doesn’t exist. If the file exists, it will be
silently overwritten, which may lead to data loss and thus requires
some caution.
ls lengths.txt confirms that the file exists:
$ ls lengths.txt
lengths.txt
We can now send the content of lengths.txt to the screen using cat lengths.txt.
cat stands for “concatenate”:
it prints the contents of files one after another.
There’s only one file in this case,
so cat just shows us what it contains:
$ cat lengths.txt
20 cubane.pdb
12 ethane.pdb
9 methane.pdb
30 octane.pdb
21 pentane.pdb
15 propane.pdb
107 total
Output Page by Page
We’ll continue to use
catin this lesson, for convenience and consistency, but it has the disadvantage that it always dumps the whole file onto your screen. More useful in practice is the commandless, which you use with$ less lengths.txt. This displays a screenful of the file, and then stops. You can go forward one screenful by pressing the spacebar, or back one by pressingb. Pressqto quit.
Now let’s use the sort command to sort its contents.
What Does
sort -nDo?If we run
sorton a file containing the following lines:10 2 19 22 6the output is:
10 19 2 22 6If we run
sort -non the same input, we get this instead:2 6 10 19 22Explain why
-nhas this effect.Solution
The
-nflag specifies a numerical rather than an alphanumerical sort.
We will also use the -n flag to specify that the sort is
numerical instead of alphanumerical.
This does not change the file;
instead, it sends the sorted result to the screen:
$ sort -n lengths.txt
9 methane.pdb
12 ethane.pdb
15 propane.pdb
20 cubane.pdb
21 pentane.pdb
30 octane.pdb
107 total
We can put the sorted list of lines in another temporary file called sorted-lengths.txt
by putting > sorted-lengths.txt after the command,
just as we used > lengths.txt to put the output of wc into lengths.txt.
Once we’ve done that,
we can run another command called head to get the first few lines in sorted-lengths.txt:
$ sort -n lengths.txt > sorted-lengths.txt
$ head -n 1 sorted-lengths.txt
9 methane.pdb
Using -n 1 with head tells it that
we only want the first line of the file;
-n 20 would get the first 20,
and so on.
Since sorted-lengths.txt contains the lengths of our files ordered from least to greatest,
the output of head must be the file with the fewest lines.
Redirecting to the same file
It’s a very bad idea to try redirecting the output of a command that operates on a file to the same file. For example:
$ sort -n lengths.txt > lengths.txtDoing something like this may give you incorrect results and/or delete the contents of
lengths.txt.
What Does
>>Mean?We have seen the use of
>, but there is a similar operator>>which works slightly differently. By using theechocommand to print strings, test the commands below to reveal the difference between the two operators:$ echo hello > testfile01.txtand:
$ echo hello >> testfile02.txtHint: Try executing each command twice in a row and then examining the output files.
Solution
In the first example with
>, the string “hello” is written totestfile01.txt, but the file gets overwritten each time we run the command.We see from the second example that the
>>operator also writes “hello” to a file (in this casetestfile02.txt), but appends the string to the file if it already exists (i.e. when we run it for the second time).
Appending Data
We have already met the
headcommand, which prints lines from the start of a file.tailis similar, but prints lines from the end of a file instead.Consider the file
data-shell/data/animals.txt. After these commands, select the answer that corresponds to the fileanimalsUpd.txt:$ head -n 3 animals.txt > animalsUpd.txt $ tail -n 2 animals.txt >> animalsUpd.txt
- The first three lines of
animals.txt- The last two lines of
animals.txt- The first three lines and the last two lines of
animals.txt- The second and third lines of
animals.txtSolution
Option 3 is correct. For option 1 to be correct we would only run the
headcommand. For option 2 to be correct we would only run thetailcommand. For option 4 to be correct we would have to pipe the output ofheadintotail -2by doinghead -3 animals.txt | tail -2 > animalsUpd.txt
If you think this is confusing,
you’re in good company:
even once you understand what wc, sort, and head do,
all those intermediate files make it hard to follow what’s going on.
We can make it easier to understand by running sort and head together:
$ sort -n lengths.txt | head -n 1
9 methane.pdb
The vertical bar, |, between the two commands is called a pipe.
It tells the shell that we want to use
the output of the command on the left
as the input to the command on the right.
The computer might create a temporary file if it needs to,
or copy data from one program to the other in memory,
or something else entirely;
we don’t have to know or care.
Nothing prevents us from chaining pipes consecutively.
That is, we can for example send the output of wc directly to sort,
and then the resulting output to head.
Thus we first use a pipe to send the output of wc to sort:
$ wc -l *.pdb | sort -n
9 methane.pdb
12 ethane.pdb
15 propane.pdb
20 cubane.pdb
21 pentane.pdb
30 octane.pdb
107 total
And now we send the output of this pipe, through another pipe, to head, so that the full pipeline becomes:
$ wc -l *.pdb | sort -n | head -n 1
9 methane.pdb
This is exactly like a mathematician nesting functions like log(3x)
and saying “the log of three times x”.
In our case,
the calculation is “head of sort of line count of *.pdb”.
Piping Commands Together
In our current directory, we want to find the 3 files which have the least number of lines. Which command listed below would work?
wc -l * > sort -n > head -n 3wc -l * | sort -n | head -n 1-3wc -l * | head -n 3 | sort -nwc -l * | sort -n | head -n 3Solution
Option 4 is the solution. The pipe character
|is used to feed the standard output from one process to the standard input of another.>is used to redirect standard output to a file. Try it in thedata-shell/moleculesdirectory!
Here’s what actually happens behind the scenes when we create a pipe. When a computer runs a program — any program — it creates a process in memory to hold the program’s software and its current state. Every process has an input channel called standard input. (By this point, you may be surprised that the name is so memorable, but don’t worry: most Unix programmers call it “stdin”). Every process also has a default output channel called standard output (or “stdout”). A second output channel called standard error (stderr) also exists. This channel is typically used for error or diagnostic messages, and it allows a user to pipe the output of one program into another while still receiving error messages in the terminal.
The shell is actually just another program. Under normal circumstances, whatever we type on the keyboard is sent to the shell on its standard input, and whatever it produces on standard output is displayed on our screen. When we tell the shell to run a program, it creates a new process and temporarily sends whatever we type on our keyboard to that process’s standard input, and whatever the process sends to standard output to the screen.
Here’s what happens when we run wc -l *.pdb > lengths.txt.
The shell starts by telling the computer to create a new process to run the wc program.
Since we’ve provided some filenames as arguments,
wc reads from them instead of from standard input.
And since we’ve used > to redirect output to a file,
the shell connects the process’s standard output to that file.
If we run wc -l *.pdb | sort -n instead,
the shell creates two processes
(one for each process in the pipe)
so that wc and sort run simultaneously.
The standard output of wc is fed directly to the standard input of sort;
since there’s no redirection with >,
sort’s output goes to the screen.
And if we run wc -l *.pdb | sort -n | head -n 1,
we get three processes with data flowing from the files,
through wc to sort,
and from sort through head to the screen.
This simple idea is why Unix has been so successful.
Instead of creating enormous programs that try to do many different things,
Unix programmers focus on creating lots of simple tools that each do one job well,
and that work well with each other.
This programming model is called “pipes and filters”.
We’ve already seen pipes;
a filter is a program like wc or sort
that transforms a stream of input into a stream of output.
Almost all of the standard Unix tools can work this way:
unless told to do otherwise,
they read from standard input,
do something with what they’ve read,
and write to standard output.
The key is that any program that reads lines of text from standard input and writes lines of text to standard output can be combined with every other program that behaves this way as well. You can and should write your programs this way so that you and other people can put those programs into pipes to multiply their power.
Redirecting Input
As well as using
>to redirect a program’s output, we can use<to redirect its input, i.e., to read from a file instead of from standard input. For example, instead of writingwc ammonia.pdb, we could writewc < ammonia.pdb. In the first case,wcgets a command line argument telling it what file to open. In the second,wcdoesn’t have any command line arguments, so it reads from standard input, but we have told the shell to send the contents ofammonia.pdbtowc’s standard input.
What Does
<Mean?Change directory to
data-shell(the top level of our downloaded example data).What is the difference between:
$ wc -l notes.txtand:
$ wc -l < notes.txtSolution
<is used to redirect input to a command.In both examples, the shell returns the number of lines from the input to the
wccommand. In the first example, the input is the filenotes.txtand the file name is given in the output from thewccommand. In the second example, the contents of the filenotes.txtare redirected to standard input. It is as if we have entered the contents of the file by typing at the prompt. Hence the file name is not given in the output - just the number of lines. Try this for yourself:$ wc -l this is a test Ctrl-D # This lets the shell know you have finished typing the input3
Why Does
uniqOnly Remove Adjacent Duplicates?The command
uniqremoves adjacent duplicated lines from its input. For example, the filedata-shell/data/salmon.txtcontains:coho coho steelhead coho steelhead steelheadRunning the command
uniq salmon.txtfrom thedata-shell/datadirectory produces:coho steelhead coho steelheadWhy do you think
uniqonly removes adjacent duplicated lines? (Hint: think about very large data sets.) What other command could you combine with it in a pipe to remove all duplicated lines?Solution
$ sort salmon.txt | uniq
Pipe Reading Comprehension
A file called
animals.txt(in thedata-shell/datafolder) contains the following data:2012-11-05,deer 2012-11-05,rabbit 2012-11-05,raccoon 2012-11-06,rabbit 2012-11-06,deer 2012-11-06,fox 2012-11-07,rabbit 2012-11-07,bearWhat text passes through each of the pipes and the final redirect in the pipeline below?
$ cat animals.txt | head -n 5 | tail -n 3 | sort -r > final.txtHint: build the pipeline up one command at a time to test your understanding
Solution
The
headcommand extracts the first 5 lines fromanimals.txt. Then, the last 3 lines are extracted from the previous 5 by using thetailcommand. With thesort -rcommand those 3 lines are sorted in reverse order and finally, the output is redirected to a filefinal.txt. The content of this file can be checked by executingcat final.txt. The file should contain the following lines:2012-11-06,rabbit 2012-11-06,deer 2012-11-05,raccoon
Pipe Construction
For the file
animals.txtfrom the previous exercise, the command:$ cut -d , -f 2 animals.txtuses the
-dflag to separate each line by comma, and the-fflag to print the second field in each line, to give the following output:deer rabbit raccoon rabbit deer fox rabbit bearWhat other command(s) could be added to this in a pipeline to find out what animals the file contains (without any duplicates in their names)?
Solution
$ cut -d , -f 2 animals.txt | sort | uniq
Which Pipe?
The file
animals.txtcontains 8 lines of data formatted as follows:2012-11-05,deer 2012-11-05,rabbit 2012-11-05,raccoon 2012-11-06,rabbit ...Assuming your current directory is
data-shell/data/, what command would you use to produce a table that shows the total count of each type of animal in the file?
grep {deer, rabbit, raccoon, deer, fox, bear} animals.txt | wc -lsort animals.txt | uniq -csort -t, -k2,2 animals.txt | uniq -ccut -d, -f 2 animals.txt | uniq -ccut -d, -f 2 animals.txt | sort | uniq -ccut -d, -f 2 animals.txt | sort | uniq -c | wc -lSolution
Option 5. is the correct answer. If you have difficulty understanding why, try running the commands, or sub-sections of the pipelines (make sure you are in the
data-shell/datadirectory).
Nelle’s Pipeline: Checking Files
Nelle has run her samples through the assay machines
and created 17 files in the north-pacific-gyre/2012-07-03 directory described earlier.
As a quick sanity check, starting from her home directory, Nelle types:
$ cd north-pacific-gyre/2012-07-03
$ wc -l *.txt
The output is 18 lines that look like this:
300 NENE01729A.txt
300 NENE01729B.txt
300 NENE01736A.txt
300 NENE01751A.txt
300 NENE01751B.txt
300 NENE01812A.txt
... ...
Now she types this:
$ wc -l *.txt | sort -n | head -n 5
240 NENE02018B.txt
300 NENE01729A.txt
300 NENE01729B.txt
300 NENE01736A.txt
300 NENE01751A.txt
Whoops: one of the files is 60 lines shorter than the others. When she goes back and checks it, she sees that she did that assay at 8:00 on a Monday morning — someone was probably in using the machine on the weekend, and she forgot to reset it. Before re-running that sample, she checks to see if any files have too much data:
$ wc -l *.txt | sort -n | tail -n 5
300 NENE02040B.txt
300 NENE02040Z.txt
300 NENE02043A.txt
300 NENE02043B.txt
5040 total
Those numbers look good — but what’s that ‘Z’ doing there in the third-to-last line? All of her samples should be marked ‘A’ or ‘B’; by convention, her lab uses ‘Z’ to indicate samples with missing information. To find others like it, she does this:
$ ls *Z.txt
NENE01971Z.txt NENE02040Z.txt
Sure enough,
when she checks the log on her laptop,
there’s no depth recorded for either of those samples.
Since it’s too late to get the information any other way,
she must exclude those two files from her analysis.
She could just delete them using rm,
but there are actually some analyses she might do later where depth doesn’t matter,
so instead, she’ll just be careful later on to select files using the wildcard expression *[AB].txt.
As always,
the * matches any number of characters;
the expression [AB] matches either an ‘A’ or a ‘B’,
so this matches all the valid data files she has.
Wildcard Expressions
Wildcard expressions can be very complex, but you can sometimes write them in ways that only use simple syntax, at the expense of being a bit more verbose.
Consider the directorydata-shell/north-pacific-gyre/2012-07-03: the wildcard expression*[AB].txtmatches all files ending inA.txtorB.txt. Imagine you forgot about this.
Can you match the same set of files with basic wildcard expressions that do not use the
[]syntax? Hint: You may need more than one expression.The expression that you found and the expression from the lesson match the same set of files in this example. What is the small difference between the outputs?
Under what circumstances would your new expression produce an error message where the original one would not?
Solution
$ ls *A.txt $ ls *B.txt- The output from the new commands is separated because there are two commands.
- When there are no files ending in
A.txt, or there are no files ending inB.txt.
Removing Unneeded Files
Suppose you want to delete your processed data files, and only keep your raw files and processing script to save storage. The raw files end in
.datand the processed files end in.txt. Which of the following would remove all the processed data files, and only the processed data files?
rm ?.txtrm *.txtrm * .txtrm *.*Solution
- This would remove
.txtfiles with one-character names- This is correct answer
- The shell would expand
*to match everything in the current directory, so the command would try to remove all matched files and an additional file called.txt- The shell would expand
*.*to match all files with any extension, so this command would delete all files
Key Points
catdisplays the contents of its inputs.
headdisplays the first 10 lines of its input.
taildisplays the last 10 lines of its input.
sortsorts its inputs.
wccounts lines, words, and characters in its inputs.
command > fileredirects a command’s output to a file.
first | secondis a pipeline: the output of the first command is used as the input to the second.The best way to use the shell is to use pipes to combine simple single-purpose programs (filters).